ادامه مطلب
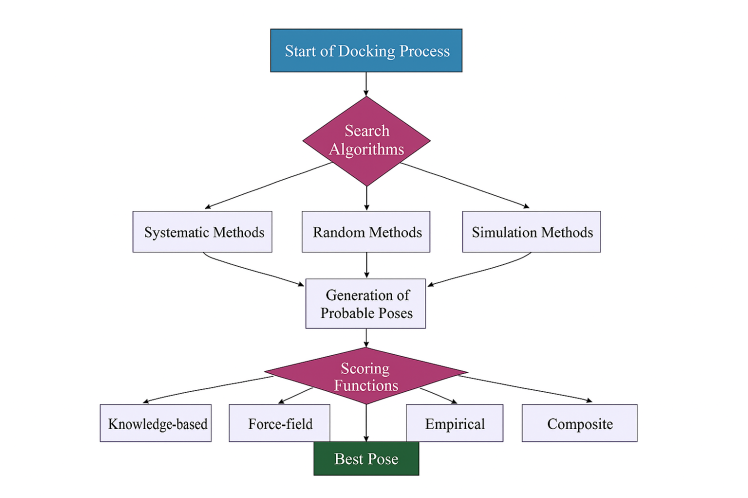
مبانی داکینگ مولکولی (Molecular Docking) - بخش ۱
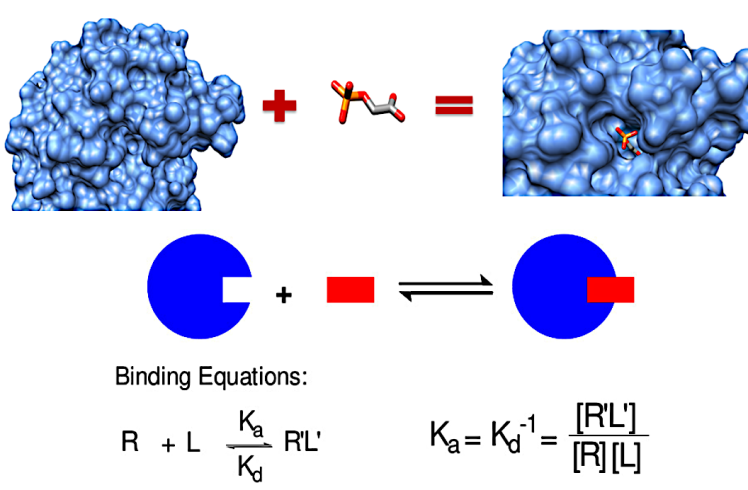
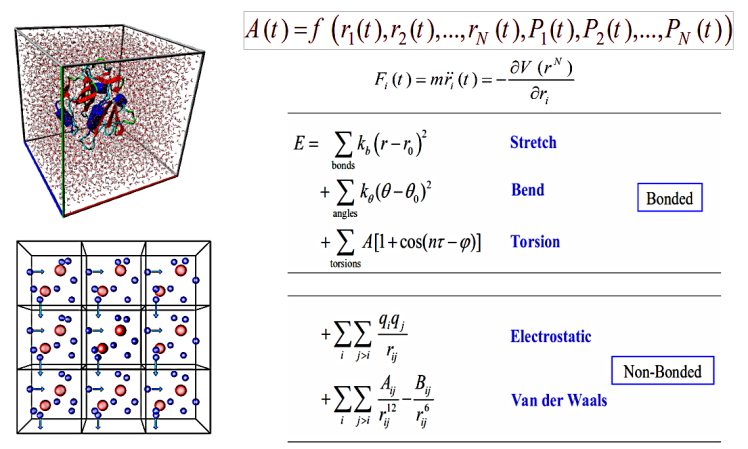
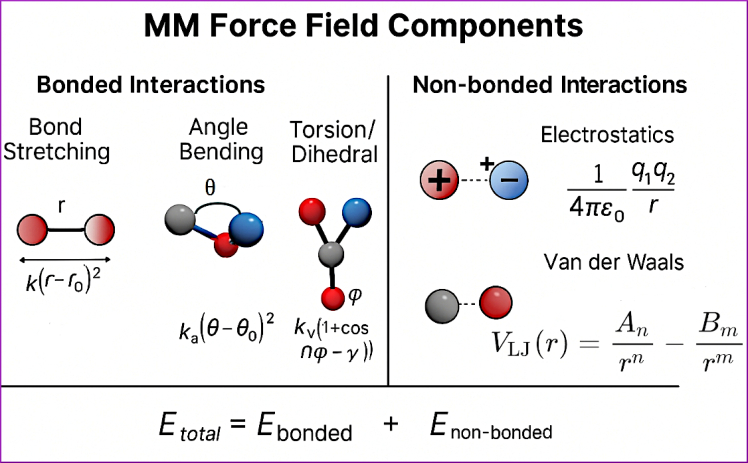
داکینگ مولکولی چیست؟ داکینگ مولکولی (Molecular Docking) یکی از روشهای مهم در زیستمحاسبات و شیمی محاسباتی است که برای پیشبینی نحوهی برهمکنش بین دو یا چند مولکول استفاده میشود. در این روش، یک مولکول کوچک مانند لیگاند در برابر یک مولکول بزرگتر مانند پروتئین، DNA یا RNA قرار میگیرد تا بهترین حالت اتصال میان آنها شناسایی شود. نتیجهی نهایی، مدلی از کمپلکس پروتئین–لیگاند است …