

مقدمهای بر یکی از مهمترین روشهای شیمی محاسباتی
فرآیندهای زیستی و شیمیایی در مقیاس اتمی و نانومتری بسیار سریع و پیچیدهاند. مشاهدهی مستقیم حرکت اتمها و مولکولها در آزمایشگاه معمولاً غیرممکن است و انجام آزمایشها در شرایط گوناگون میتواند پرهزینه یا حتی غیرعملی باشد.
در چنین شرایطی، شبیهسازی دینامیک مولکولی (Molecular Dynamics Simulation) به عنوان یکی از قدرتمندترین ابزارهای شیمی محاسباتی وارد عمل میشود.
در این روش با استفاده از قوانین فیزیک کلاسیک، رفتار اتمها و مولکولها در طول زمان مدلسازی میشود. به کمک این شبیهسازیها میتوان مسیر حرکت ذرات، برهمکنشهای درونمولکولی و بینمولکولی، و تغییرات انرژی را با دقت بالا بررسی کرد.
بهبیان ساده، شبیهسازی دینامیک مولکولی یعنی ساخت یک «جهان میکروسکوپی مجازی» در کامپیوتر، تا بتوانیم رفتار سیستمهای واقعی را پیشبینی و تحلیل کنیم.
از دیدگاه میکروسکوپی تا خواص ماکروسکوپی
یکی از اهداف اصلی در شبیهسازی مولکولی این است که از رفتار ذرات کوچک، بتوان خواص کلی و بزرگ مقیاس ماده را پیشبینی کرد. این ارتباط میان دو مقیاس با استفاده از مکانیک آماری (Statistical Mechanics) برقرار میشود.
در مکانیک آماری، خواص ماکروسکوپی مثل دما، فشار، انرژی یا چگالی از روی حرکت ذرات محاسبه میشود.
بههمین دلیل، وقتی در شبیهسازی دینامیک مولکولی حرکت تکتک اتمهای یک پروتئین یا مولکول را دنبال میکنیم، میتوانیم رفتار کلی آن سیستم را تحلیل کنیم، مثلاً پایداری ساختار، برهمکنش با داروها یا نحوهی تغییر شکل در دماهای مختلف.
روشهای شبیهسازی مولکولی
بهطور کلی دو روش اصلی برای شبیهسازی سیستمهای اتمی وجود دارد:
۱. شبیهسازی مونتکارلو (Monte Carlo Simulation)
شبیهسازی مونتکارلو یک روش تصادفی است که با نمونهبرداری هوشمند از پیکربندیهای مختلف یک سیستم، بهطور خاص برای مطالعه حالت تعادل ترمودینامیکی و محاسبه خواص آماری سیستمها (مانند انرژی آزاد) استفاده میشود. این روش بر خلاف دینامیک مولکولی، مسیر زمانی واقعی را دنبال نمیکند
۲. شبیهسازی دینامیک مولکولی (Molecular Dynamics – MD)
در این روش، معادلات حرکت نیوتن برای تمامی ذرات حل میشود تا مسیر حرکت (Trajectory) آنها در طول زمان بهدست آید.
دینامیک مولکولی اطلاعات بسیار دقیقتری دربارهی زمان، انرژی و ساختار سیستم فراهم میکند و پلی میان ساختار مولکولی و عملکرد زیستی یا فیزیکی آن ایجاد میکند.
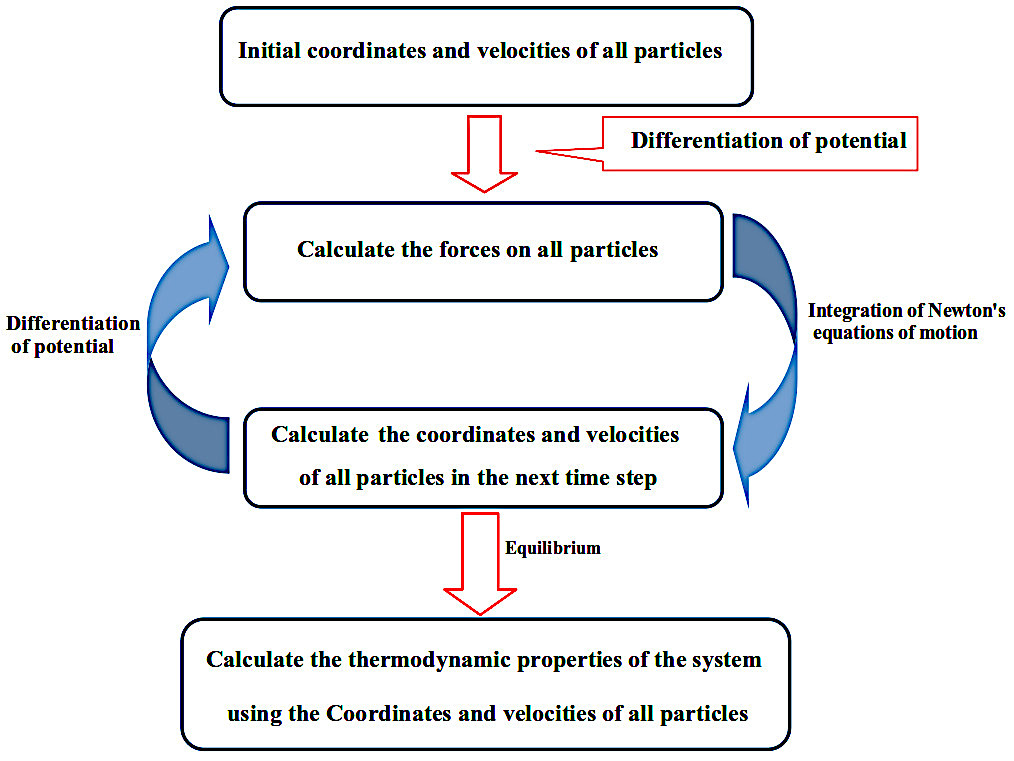
مراحل انجام شبیهسازی دینامیک مولکولی
۱. تعیین شرایط اولیه:
موقعیت و سرعت اولیهی اتمها مشخص میشود. این دادهها معمولاً از ساختارهای کریستالوگرافی یا فایلهای PDB استخراج میشوند.
۲. محاسبهی نیروها:
با استفاده از تابع انرژی پتانسیل، نیروهای وارد بر هر ذره محاسبه میشود.
این تابع شامل برهمکنشهای درونمولکولی (پیوندها، زاویهها، چرخشها) و برهمکنشهای بینمولکولی (نیروهای واندروالسی، الکترواستاتیکی، پیوندهای هیدروژنی و π–π interactions) است.
۳. حل معادلات حرکت نیوتن:
با استفاده از نیروهای محاسبهشده، موقعیت و سرعت جدید اتمها در گام زمانی بعدی بهدست میآید.
گام زمانی معمولاً در حد فمتوثانیه (10⁻¹⁵ ثانیه) است تا دقت حرکتهای اتمی حفظ شود.
۴. تکرار و تولید تراژکتوری:
مراحل فوق میلیونها بار تکرار میشوند تا مسیر زمانی سیستم (Trajectory) بهدست آید. پس از رسیدن سیستم به تعادل، میتوان خواص ترمودینامیکی را از این مسیر استخراج کرد.
درک فیزیکی از فرایند شبیهسازی
در مکانیک آماری یک قانون کلیدی وجود دارد:
اگر تراژکتوری (Trajectory) یک سیستم را داشته باشیم، یعنی بدانیم موقعیت و سرعت ذرات در هر لحظه چگونه تغییر میکنند، میتوانیم همهی خواص ترمودینامیکی آن سیستم را محاسبه کنیم.
در شبیهسازی دینامیک مولکولی دقیقاً از همین اصل استفاده میشود. در ابتدا موقعیتها و سرعتهای اولیهی ذرات تعیین میشود. سپس از روی مدل پتانسیل انرژی، نیروهای وارد بر تکتک ذرات با مشتقگیری محاسبه میگردد. در ادامه با کمک انتگرالگیری از معادلات حرکت نیوتن، موقعیتها و سرعتهای جدید برای گام زمانی بعدی بهدست میآیند.
این چرخه (محاسبه نیرو با مشتق گیری از پتانسیل، انتگرالگیری از معادلات حرکت نیوتن، بهروزرسانی موقعیت و سرعت) میلیونها بار تکرار میشود تا سیستم به تعادل ترمودینامیکی برسد. در این حالت، کمیتهای فیزیکی حول مقدار میانگین خود نوسان میکنند. دقیقاً به دلیل تکرار همین چرخۀ محاسباتی سنگین است که برای اجرای شبیهسازیهای دینامیک مولکولی به کامپیوترها و سرورهای بسیار پرسرعت و غالباً اَبَررایانهها نیاز داریم.
وقتی سیستم به تعادل میرسد، تراژکتوری کامل آن ثبت شده است. از این دادهها میتوان با میانگینگیری و روابط آماری، کمیتهای ترمودینامیکی را محاسبه کرد.
کاربردهای شبیهسازی دینامیک مولکولی
جمعبندی
شبیهسازی دینامیک مولکولی ابزاری قدرتمند برای درک رفتار سیستمهای مولکولی در سطح اتمی است.
این روش، با ترکیب قوانین فیزیک کلاسیک، مدلسازی دقیق انرژی و توان محاسباتی رایانهها، پلی میان دنیای واقعی و دنیای محاسباتی ایجاد میکند.
در بخش ۲ از مجموعهی «مبانی شبیهسازی دینامیک مولکولی» به بررسی انواع مدلهای پتانسیل انرژی خواهیم پرداخت و خواهیم دید که چگونه از دادههای خام شبیهسازی، اطلاعات فیزیکی دقیق استخراج میشود.
پیشنهاد ویژه
در مرکز پژوهشهای رایانهای ایلیا ما تلاش میکنیم با ارائهی آموزشهای تخصصی، شبیهسازیهای علمی و تحلیل دادهها در زمینهی نرمافزارهای محاسبات مولکولی، به پژوهشگران کمک کنیم تا درک عمیقتر و دقیقتری از دینامیک سیستمهای مولکولی بهدست آورند.
آموزش مقدماتی مبانی شبیه سازی دینامیک مولکولی